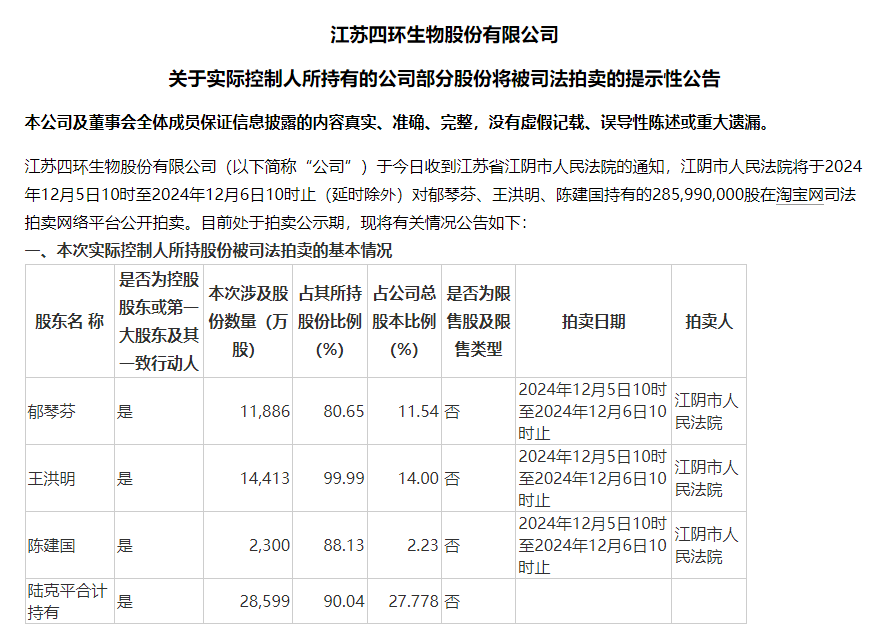
40余款全球前沿创新产品登陆进博医疗创新孵化专区,全球合力共筑产业创新生态
上海2024年11月9日 /美通社/ — 11月4日至5日,由进博展盟医疗器械专委会(以下简称"专委会")及万疆创新共同主办的万疆国际医疗创新暨进博医疗创新孵化研讨会(以下简称"研讨会")如期召开。来自国家药监局、地方政府、跨国医械企业及投资机构等领域重磅嘉宾齐聚一堂,共同探讨全球医疗器械领域发展现状与未来机遇。此外,专委会还携手万疆创新及相关政府单位,共同举办了国际医疗创新产品路演活动,来自瑞士、意大利、法国等7个国家的共计12款创新展品在路演中亮相。
在为期两天的时间里,研讨会不仅汇集众多行业精英与业界领袖,就医疗器械领域前沿话题分享了他们的深入洞见和前瞻思考,还通过创新产品路演活动助力加速全球顶尖医疗创新成果落地中国,合力共筑高质量发展、具有鲜明中国特色的全球开放式融合创新的医疗科技产业生态。
进博会展商联盟医疗器械专业委会会长单位代表、碧迪医疗全球高级副总裁、大中华区总经理邓建民在主旨演讲中表示,"中国医疗器械行业已砥砺前行30年,从仿制创新走向变革性创新,逐步迈入高质量发展的新纪元。创新孵化专区作为我国医疗领域创新资源与全球接轨的开放平台,为全球医疗创新企业与本土创新的深度融合提供动力。碧迪医疗作为医疗器械专委会会长单位,将继续携手专委会成员单位等各方合作伙伴,为国内外医疗创新企业搭建开放交流与寻求合作的平台,共同推动医学科技的进步与发展。"
把握政策红利,激励全球医疗创新企业"走进来"
在人口老龄化带来的医疗需求不断增长、我国积极吸引外资与先进技术进入中国的大背景下,全球前沿医疗创新技术成果落地中国已成必然趋势。随着国际医疗交流与合作日益频繁,全球医疗行业创新链、产业链、资金链及人才链也趋向深度融合。在此背景下,越来越多来自海外的创新医疗企业希望抓住高水平开放的"中国机遇",借助进博平台将其创新医疗技术成果引入中国,并期待在中国市场深耕发展。
然而,许多海外企业对中国医疗器械市场准入与注册、市场监管及知识产权保护等政策法规存在知识盲区。为了加深跨国医疗企业对中国市场准入与监管相关政策法规的了解,激励更多医疗创新企业"走进"中国市场,研讨会特别围绕"知识产权保护""市场监管与产品准入"等话题开展了精彩的主题分享和圆桌讨论。这些内容与今年虹桥国际经济论坛"保护知识产权,加强国际打击侵权假冒合作"等核心议题高度契合,彰显了研讨会与国家政策的紧密契合,为海外创新成果在中国市场的顺利落地与转化提供了宝贵的策略建议与支持服务。
知识产权作为技术创新与市场竞争的核心要素,在医疗器械这一技术高度密集型行业中具有举足轻重的作用。研讨会期间,在万疆创新合伙人、上海瓴德律师事务所创始人与管理合伙人林忠博士的主持下,河南省知识产权局局长刘怀章先生、瑞士医疗科技公司Terapet首席执行官兼联合创始人Christina Vallgren女士、Lenz & Staehelin合伙人与知识产权部负责人Sevan Antreasyan先生、瓴德律师事务所合伙人李雨擎女士和碧迪医疗法务顾问林松先生共同围绕中国知识产权保护的最新进展进行了深入讨论,为来自全球的创新医疗企业如何加速产品引入指点迷津。
得益于一系列推动医疗器械行业创新与发展的政策扶持,中国已成功跃居为全球第二大医疗器械市场。与此同时,中国正不断优化和完善医疗器械监管框架,对产品的安全性、有效性及创新性设立了更为严苛的标准。在科技进步日新月异的背景下,以及浓厚的创新氛围驱动下,中国本土医疗器械企业的创新能力显著提升。面对国内外竞争环境的日益白热化,中国医疗创新领域正步入一个机遇与挑战并存的高质量发展新时期。在此背景下,波士顿科学市场准入部门负责人周践女士、泰格捷通总经理彭沂非先生、信迈医疗首席执行官王捷博士,以及杭州泰源医药创新研究院副总经理高松博士,在研讨会上就创新医疗科技产品在中国市场的准入机遇和挑战进行了深入的交流与探讨。
聚焦行业前沿,共话医疗产业未来创新生态圈
在全球医疗领域的国际交流与合作日趋频繁的背景下,研讨会为来自全球各个国家和地区的医疗创新企业提供了展示前沿产品和寻求投资合作的开放平台。中国的营商环境持续优化,展现出了前所未有的活力与吸引力,为全球医疗企业创造了巨大的市场机遇。会上,华瑭大昌首席执行官程学蕙、瑞士中心首席执行官肖振、瑞士贸易与投资署医疗科技负责人Nicolas Panzer和威高集团副总裁、威高骨科董事长陈敏共同探讨了中欧之间在医疗领域的合作和协同创新的可能潜力。此外,会议还特别邀请了Denovo Solutions首席执行官Alaeddin Ahram分享中东市场的创新医疗科技机遇,为与会者带来了全新国际合作视野。
瑞士中心集团首席执行官肖振在讨论中表示,"瑞士中心作为进博会官方主展机构之一,一直致力于为瑞士中小企业进入中国市场发挥桥梁作用。而进博创新孵化专区的设立,不仅为许多瑞士小微企业提供了展示自身创新潜力的机会,还提供了一个寻求投资和合作的交流平台。未来,我们希望能继续借助创新孵化专区,持续引进来自瑞士医疗器械企业的创新产品与技术,并助推更多来自瑞士的医疗创新技术在中国市场生根发芽,进一步促进中瑞企业在医疗器械领域的交流与合作。"
针对中国当前医疗领域的投资现状和前景,华平投资中国区联席CEO方敏也在会上发表了深刻洞见:"中国医疗器械市场的增长非常迅速,但目前市场还未被充分渗透,人口老龄化的加深也为全球先进医疗技术及医疗设备带来了巨大的市场潜力和需求。在创新研发方面,中国在医疗器械的创新研发支出及风险投资仅次于美国,位列全球第二。近年来,政府也在不断出台激励和扶持政策,多方面因素共同推动了中国医疗器械市场的创新浪潮,中国医疗器械市场更是成为了全球投资瞩目的重要赛道。我们非常荣幸能够借进博医疗创新孵化专区这一开放平台,接触到全球领先的医疗创新科技,也希望可以通过资本的力量,加速海外创新项目在中国的落地。"
深化在华战略,分享本土化成功"范本"
11月4日,碧迪医疗与河南省政府在研讨会期间举办了战略合作揭牌仪式,女用导尿包创新项目成果实现本土化落地,也标志着双方正向着更深一层的合作迈进。对此,河南省政府副秘书长周峰表示:"多年来,河南省深入实施创新驱动、科教新省、人才强省战略,高水平规划建设中原医学科学城,致力于建设医学科学创新高地,推动全省生物医药大健康产业跨越式发展。未来,河南将进一步加强与世界各国企业、科研机构和专家学者的交流合作,不断扩大国际医疗健康朋友圈,持续打造市场化、法治化、国际化的一流营商环境,助力医疗健康领域龙头企业在河南大展宏图,为健康中国建设贡献更多力量。"
会上,来自罗氏诊断、西门子、碧迪和威高骨科等多名跨国药械企业代表分享其在医疗创新、战略合作等本土化成功经验,为参会者提供了宝贵的本土化实践借鉴。其中,罗氏诊断中国区总经理姚国樑先生以"开拓新领域:推动中国诊断技术与数字健康的创新"为主题,分享了罗氏聚焦中国健康事业发展需求,引领并赋能医疗行业发展的故事。此外,碧迪医疗全球高级副总裁、大中华区总经理邓建民先生分享了与河南省政府、河南亚都控股集团就全球首创PUREWICK®女用导尿棒进行的本土战略合作,并与快奥森多大中华区总裁林妍女士还围绕"本土制造战略与实践"开展了深度对话,为与会的海外企业提供了跨国本土化战略布局的深刻见解。
紧跟创新前沿,助力优秀创新成果落地"孵化"
11月5日,专委会还携手万疆创新、天津市政府和瑞士中心等相关单位,共同举办了国际医疗创新产品路演活动,让更多中国医疗企业更深入了解来自海外的创新医疗技术,促进中外医疗企业深入交流与合作。路演期间,来自瑞士、意大利、法国等7个国家的共12款创新展品成功亮相。
天津市市委常委、河西区委书记王旭在研讨会路演专场的开场致辞中表示:"天津一直以来都高度重视健康产业发展,并不断加快推动‘天津基地'建设,携手打造医疗健康领域科技创新、医疗服务、人才培养和成果转化新高地。未来,天津也将持续优化营商环境,以开放的态度欢迎全球各个国家和地区的医疗创新企业投资天津,借助进博会及医疗创新孵化研讨会这一开放交流平台,助推更多全球前沿医疗技术成果落地天津,从而推动全球医疗器械产业的高质量发展。"
在参加路演活动的创新展品中,有不少创新技术和产品均实现了跨领域、跨学科式融合创新,为与会的中国医疗企业带来了一场"创新盛宴"。其中,在肿瘤领域,由瑞士医疗科技公司Terapet开发的核医学成像设备Qualyscan®,是全球首款用于癌症治疗的新型质子疗法,能够确保每位患者每次都能获得精确的剂量。该产品能够以非侵入式的方式,在质子治疗全过程中对患者体内的质子剂量进行实时监测和控制,从而提升质子治疗的精准度、安全性与可及性。
在医美和眼科领域,由意大利GMV公司潜心研发的等离子皮肤及眼科治疗装置Plexr®采用全新非侵入性的等离子强效抗衰除皱技术,能够达到不用手术刀就能去除松弛多余皮肤的效果,从而为广大追求眼部年轻化的爱美人士提供了全新选择。由STAAR Surgical研发的EVO+ Visian ICL采用Collamer®专利材料制成,用于矫治近视和散光,旨在为近视患者带来更高清、更舒适的视觉体验。
在专委会的牵头下,来自全球各地的企业创始人和投资人等多名行业专家,也凭借其对海外创新生态和中国市场的深入洞察,对参加路演的产品给予了评价与反馈,并把全球优秀的最新医疗创新产品推荐给了中国医疗行业,助力小微创新企业圆梦中国。
40余款全球前沿创新产品登陆进博医疗创新孵化专区
进博期间,进博医疗创新孵化专区展台特设"瑞士中心专区"和"精品专区",联合展示40余款来自全球企业的医疗创新技术及产品,涵盖了医美整形、眼科医疗、体外诊断、呼吸系统疾病、血液透析以及心血管健康等多个专业领域。得益于瑞士中心的鼎力支持,专区展台设计也巧妙融入了瑞士特色元素,旨在彰显中欧医疗技术的紧密交流与深度合作。
自2021年起,碧迪医疗已连续四年荣膺进博会展盟医疗器械专委会会长单位身份,并领衔43家会员企业助力进博会医疗展区设立创新孵化专区。三年来,中国不断优化的营商环境和创新氛围,持续吸引全球领先的医疗科技企业深化在中国的布局,拓展本土战略。作为面向全球领先医疗科技的国际开放窗口和孵化沃土,专区已累计从全球多项创新项目中筛选后成功引进和展示创新成果147项,其中实现中国本土运营18项,9项推进战略授权合作并实现商业化。
进博医疗创新孵化专区始终以患者需求为核心,致力于带来可持续、高价值、定制化的全球前沿创新产品、服务和解决方案。在发展新质生产力、促进高水平开放的时代要求和政策背景下,进博医疗创新孵化专区不仅致力于引进海外先进的医疗技术和创新产品,积极推动全球前沿医疗创新成果落地中国,还为国内医疗企业提供了科技交流和海外市场合作的机会,促进国内外医疗创新技术的交流和融合,以实际行动践行"赋能共创 圆梦东方"的崇高理念,为推动构建人类卫生健康共同体贡献力量。
未来,专区将持续引进全球最先进、最具影响力的医疗技术创新成果,为全球优质小微企业提供试验场和孵化器,加速促进医疗技术产业发展与转型升级。专委会也将持续提升医疗创新孵化专区的价值和影响力,力争把医疗创新孵化专区打造成具有全球影响力的医疗创新平台和孵化沃土,助力健康中国2030宏伟目标的实现。